cell-biology
یه جا برای یاد دادن و یاد گرفتنcell-biology
یه جا برای یاد دادن و یاد گرفتنکلونینگ ژن
ژن کلونینگ
ژن کلونینگ فرآیندی است که طی آن توالی مشخصی از DNA را جداسازی میکنند تا نسخههای یکسانی از آن را در محیط طبیعی ( سلول یا بافت زنده ) بدست آورند.
هدف از ژن کلونینگ فراهم کردن نسخههای متعدد از یک ژن منفرد است. تکثیر یک ژن در حوزههای مختلف تحقیقاتی مورد استفاده است. به علاوه دارای کاربردهای پزشکی از قبیل ژن درمانی و کاربردهای صنعتی نظیر تولید مقدار زیادی از یک پروتئین میباشد.
برای کلون کردن ژن قطعهای از DNA را از موجودی به موجود دیگر منتقل میکنند. سلولی را که منشا DNA از آن است را « دهنده » و سلولی را که آن را دریافت میکند « میزبان» میگویند.

کلونینگ ژن به روشهای مختلفی صورت میگیرد اما اساس همهی آنها به این صورت است که DNA هدف از سلول دهنده استخراج میشود و با کمک آنزیمهایی برش داده میشود و به داخل یک مولکول DNA حلقوی، که معمولاً یک پلاسمید است و نقش ناقل ( وکتور ) را دارد، وارد میشود. به این ترتیب یک مولکول DNA نوترکیب ساخته میشود. در مرحلهی بعد DNA نوترکیب را به سلول میزبان که اغلب نوعی باکتری میباشد منتقل میکنند. این مرحله را ترنسفورماسیون مینامند. DNA نوترکیب در سلول میزبان همانندسازی میکند و همراه سلول میزبان تکثیر میشود. سلولهای حاصل از تقسیم سلول میزبان اولیه، نسخههایی از DNA نوترکیب همانندسازی شده را به ارث میبرند. سلولهای باکتری به دنبال تقسیمات متعدد کلنی تشکیل میدهند و از آنجا که اعضای این کلونی حاوی یک یا چند نسخه از ژن مورد نظر ما که در DNA نوترکیب حمل میشود میباشد، میتوان گفت این ژن کلون شده است.
استخراج و برش ـ اولین گام در تولید DNA نوترکیب و سپس کلون کردن آن، استخراج و برش وکتور از سلول باکتری و DNA از سلول دهنده میباشد.
برای استخراج DNA از سلول دهنده، ابتدا باید «عصارهی سلولی» تهیه کرد. برای این منظور غشای سلول را متلاشی میکنند تا محتویات آن خارج شود سپس مخلوط حاصل را سانتریفیوژ میکنند تا زوائد آن تهنشین شود و مایع رویی که «عصارهی سلولی» است و حاوی DNA میباشد را جمع آوری میکنند. عصارهی سلولی علاوه بر DNA حاوی پروتئین و RNA نیز میباشد. با یکی از روشهای «تجزیهی آنزیمی پروتئین و RNA » یا «کروماتوگرافی تعویض یونی» DNA را از عصارهی سلولی تخلیص میکنند.
برای استخراج پلاسمید از سلول باکتری، بعد از تخلیص DNA باید پلاسمید را از DNA کرومزوم باکتری جدا کرد. برای این منظور از تکنیک « اولتراسانتریفیوژ » که بر مبنای شیب جرمی میباشد استفاده میکنند.
بعد از جداسازی، DNA سلول دهنده باید به قطعات کوچکتر بریده شود. برش DNA به قطعات کوچکتر را میتوان از طرق راههای مختلفی انجام داد از آن جمله ایجاد شکست در DNA با استفادها از امواج صوتی ( سونیکیت )میباشد. در این صورت طول قطعات حاصل از یک مولکول DNA با مولکول دیگر متفاوت خواهد بود چرا که شکست مولکول DNA به صورت تصادفی صورت میگیرد. کشف «آنزیمهای محدود کننده» (restriction enzymes) محققان را قادر ساخت تابتوانند قطعات با طول یکسان را از چندین مولکول DNA یکسان فراهم کنند. آنزیمهای محدود کننده باکتریها را قادر به دفاع در مقابل فاژها میکند. این آنزیمها مولکول DNA را درمحلهای ویژهای باترتیب نوکلوتیدی خاصی قطع میکنند. بنابراین برای آنکه یک مولکول DNA با این آنزیمها بریده شود، وجود توالیهای خاصی به نام «جایگاه محدود کننده» در مولکول DNA ضروری است و باید برای برش DNA به دنبال آنزیمی گشت که دارای بیش از 2 جایگاه محدود کننده بر روی آن باشد.
همچنین مزیت دیگر استفاده از آنزیمهای محدود کننده این است که میتوان از همان آنزیم برای برش پلاسمید ناقل استفاده کرد و به این طرق امکان انطباق دو انتهای قطعهی ژن هدف با دو انتهای بریده شدهی پلاسمید را به سادگی فراهم کرد.

استخراج قدیمی ترین DNA
دانشمندان موفق به استخراج DNA نئاندرتالی که 100 هزار سال قبل میزیست شدهاند. این قدیمیترین DNA انسانی است که تاکنون کشف میشود.
 این DNA از دندان بقایای کودکی از نسل نئاندرتال ها که در غار اسکلادینا در آبگیر رود میوز در بلژیک پیدا شده بود استخراج شده است.
این DNA از دندان بقایای کودکی از نسل نئاندرتال ها که در غار اسکلادینا در آبگیر رود میوز در بلژیک پیدا شده بود استخراج شده است.این مطالعه که نتایج آن در نشریه Current Biology گزارش شده است، حاکی از آن است که خویشاوندان دوردست ما از لحاظ ژنتیکی متنوعتر از آن بودند که قبلا تصور میشد.
البته زمانی که پای انسان های اولیه در حدود 35 هزار سال قبل به اروپا باز شد، از تنوع ژنتیکی نئاندرتال ها، شاید به خاطر تغییرات آب و هوایی یا بیماری ها، به شدت کاسته شده بود.
پژوهشگران فرانسوی و بلژیکی این مواد ژنتیکی را از میتوکندری جدا کردند.
دانشمندان توالی 123 جفت باز DNA را رمزگشایی کرده و آن را با سایر توالیهای DNA نئاندرتال هایی به قدمت 29 هزار سال تا 42 هزار سال مقایسه کردند.
گروهی فرانسوی تحت سرپرستی دکتر کاترین هانی در این نشریه نوشت: "توالی اسکلادینا آشکار کرده است که تنوع ژنتیکی نئاندرتال ها تاکنون کمتر از حد واقعی تخمین زده شده بود."

این یافته ها حکایت از آن دارد که تنوع ژنتیکی در دوره های ابتدایی تاریخ نئاندرتالها بیشتر از زمانهای اخیرتر، یعنی دوران آغاز ورود انسان به اروپا، بود.
نئاندرتالها از 230 هزار تا 28 هزار سال قبل در اروپا، آسیای میانه و خاور میانه میزیستند.
آنها شکارچیانی ماهر بودند و به خوبی خود را برای زندگی در عصر یخبندان تطبیق داده بودند؛ اما پس از ظهور انسان های مدرن (کرو-مگنون ها) به تدریج از صفحات اروپا محو شدند.
علت انقراض نئاندرتالها مجهول است، اما نظریه های گوناگونی در این زمینه مطرح شده است که از جمله عوامل زیستی، زیست محیطی و فرهنگی را ذکر می کند.
مطالعاتی که تا به امروز روی DNA صورت گرفته حاکی از آن است که انسان و نئاندرتالها به ندرت با یکدیگر آمیزش کرده یا هرگز چنین نکردهاند.
منبع:BBC
ژن های کاذب
ژنهای کاذب (Pseudogenes) توالیهایی مرتبط با ژنهای شناخته شده هستند که تونایی کد کردن پروتئین (یا تولید رونوشت RNA) را از دست دادهاند. بنا بر این میتوان گفت که ژنهای کاذب بر اساس دو خصوصیت تشابه (Homology) با ژنهای عملکردی و عدم فعالیت مشخص میشوند. ژنهای کاذب بر اساس مکانیسم به وجود آمدنشان به چند گره تقسیم میشوند:
1- ژنهای کاذب پیراسته (Processed) یا رونویسی معکوس شده (Retrotransposed) که حاصل رونویسی معکوس از روی RNA هستند (cDNA). از آنجا که mRNA فاقد اینترون و توالیهای بالادستی (Promoter and ...) است این دسته از ژنهای کاذب نیز فاقد این توالیها هستند.
2- ) کاذبNon-processedژنهای ناپیراسته (یا مضاعف شده (Duplicated) محصول نوآراییهایی (Recombinations) هستند که سبب مضاعف شدن (Duplication) ژنها میشوند. پس از مضاعف شدن، ممکن است یکی از نسخهها به علت بروز جهش نقطهای (Point-mutations) غیر فعال گشته وتبدیل به ژن کاذب گردد. این دسته از ژنهای کاذب دارای ساختار کامل ژن هستند (پروموتر، اینترونها و ...).
3- (Disabled genesژنهای ازکارافتاده( یا ژنهای کاذب یگانه (Unitary) حاصل غیر فعال شدن یک ژن عملکردی هستند. غیر فعال شدن توسط همان مکانیسمهایی که در ایجاد ژنهای کاذب ناپیراسته دخیل هستند رخ میدهد، اما در این گروه مضاعف شدن صورت نمیگیرد و تنها نسخه موجود از ژن غیرفعال میشود. ژن ازکارافتاده ممکن است تحت تاثیر انتخاب طبیعی در جمعیت باقی مانده و پایدار شود. به عنوان مثال میتوان به ژن GLO (L-gulono-γ-lactone oxidase) که در اکثر پستانداران در بیوسنتز اسکوربیک اسید نقش دارد اما در انسان و سایر نخستیها (Primates) تنها یک نسخه غیر فعال از آ وجود دارد.

برخی از ژنهای کاذب ممکن است به نحوی فعالیت خود را بازیابند. این ژنها را ژنهای کاذب عملکردی (Functional pseudogenes) گفته میشود. این مسئله ممکن است تحت تاثیر پروموتر ژن مجاور یا فعال شدن پروموتر خود ژن کاذب رخ دهد. رونویسی از ژنهای کاذب عملکردی معمولاً مختص به بافت (tissue-specific) است. مواردی از فعالیت رونوشت ژن کاذب پیراسته به عنوان تنظیمگر ترانس (trans-regulatory) برای ژن اصلی مشاهده شده است. رونویسی از ژنهای کاذب عملکردی معمولاً مختص به بافت (tissue-specific) است. مواردی از ژنهای کاذب پیراسته که پروتئین عملکردی تولید میکنند و همچنین مواردی از siRNA تولید شده از این ژنها شناسایی شده است.
جهت مطالعه بیشتر به www.pseudogene.org مراجعه کنید.
منبع :
http://molbio.persianblog.ir
ژن درمانی راهی به سوی پیشرفت پزشکی
معرفی:
خاموش کردن اختصاصی بیان ژنها توسط فرآیند تداخل RNA (RNA interference) یا RNAiیک مکانیسم تنظیم بیان ژنها میباشد که در سطح پس از نسخهبرداری در سلولهای یوکاریوتی اعمال میشود. این پدیده که توسط مولکولهای دو رشتهای RNA بنام مولکولهای RNA مداخلهگر کوچک (small interfering RNA) یا siRNA واسطهگری میشود از لحاظ تکاملی در میان موجودات یوکاریوت حفظ شده است و بنظر میرسد که جهت حفاظت ژنوم در مقابل تهدیدات ژنهای با منشاء خارجی (اگزوژن) نظیر ژنهای ویروسی وترانسژنها و همچنین ژنهای با منشاء داخلی نظیر عناصر ژنتیکی متحرک مثل ترانسپوزونها بکار میرود. علاوه بر این فرآیند تداخل RNA در برنامههای سلولی جهت تنظیم بیان ژنها و کنترل رشد و نمو سلولی نقش دارد.
تاکنون سه شکل از فرآیند تداخل RNA که از لحاظ فنوتیپی متفاوت ولی از جهت مکانیسم عمل یکسان و مشابهاند در یوکاریوتها شناسایی شده است. این اشکال شامل همسرکوبی (Cosuppression) یا خاموش شدن ژن پس از نسخهبرداری
(Post-Transcriptional Gene Silencing) یا PTGS در گیاهان، فرونشانی ژنها (Quelling) در قارچها و تداخل RNA (RNA interference) یا RNAi در سلسلهی جانوری میباشند.
تاریخچه:
پدیده خاموشی ژن با واسطه RNA دو رشتهای (dsRNA-mediated gene silencing) نخستین بار در گیاهان در سال 1990 توسط Napoli و همکاراناش بطور اتفاقی طی تحقیقی بر روی گلهای اطلسی ترانسژن (transgenic petunia) کشف گردید. در این بررسی دانشمندان با استفاده از ورود ترانسژنهای اگزوژن به درون سلولهای گیاه، گیاهان نوترکیبی را تولید کردند به این امید که میزان پیگمانتاسیون و تولید رنگدانه را افزایش دهند. اما با ورود ترانسژنهای اگزوژن نتیجه مورد انتظار حاصل نشد به گونهای که بسیاری از گلها دارای فنوتیپ رنگارنک و بعضی بدون رنگدانه بودند وگلهای بیرنگ بجای ارغوانی حاصل شد. متعاقب این مطالعات واژهی همسرکوبی (Cosuppression) برای توصیف خاموشی همزمان بیان mRNA های اندوژن و اگزوژن توسط Jorgenson معرفی شد. بعدها پدیدههای مشابهی در قارچها، حشرات، انگلها، موش و ردههای سلولی انسانی گزارش شد. در سال 1997 Cogoni و Macino پدیده مشابهی را در قارچ نوروسپورا کراسا (Neurospora crassa) مشاهده کردند و آن را یک سازوکار دفاعی ضد ویروسی از سوی قارچ دانستند. در قارچ نوروسپورا کراسا پدیده همسرکوبی ژنها با عنوان فرونشانی (Quelling) نامیده میشود. با این وجود کشف قطعی پدیده تداخل RNA زمانی صورت گرفت که Andrew Fire و Craig Mello سرگرم توجیه خاموشی غیر منتظره ناشی از حضور RNA سنس (senseRNA) بودند و در پی آن در سال 1998 نقش حیاتی مولکول RNA دو رشتهای در خاموش شدن اختصاصی توالی ژنها در نماتود کانورابدیتیس الگانس (Caenorhabditis elegans) تشخیص داده شد. فایر و همکاراناش موفق به اثبات این موضوع شدند که به هنگام هدف قرار دادن اختصاصی نسخه mRNA یک ژن در نماتود C. elegans، تزریق مولکولهای dsRNA قادر است حداقل تا ده برابر اثر خاموشی قویتری را در مقایسه با مولکولهای senseRNA و یا antisenseRNA به تنهایی القا کند. این مشاهده منجر به حصول این نتیجه شد که تزریق dsRNA اختصاصی با منشاء خارجی به بدن نماتود به گونهای بسیار کارآمد سبب پاسخ خاموشی اختصاصی ژن هدف در بدن این موجود میشود. این پدیده نوظهور، تداخل (RNA interference) RNA یا همان RNAi نامیده شد. این پدیده در دیگر موجودات یوکاریوتی اعم از جانوران، گیاهان و انسان به خوبی مورد مطالعه قرار گرفت و سازوکارهای آن شناخته شده است. هم اکنون، تکنیک تداخل RNA به عنوان ابزاری توانمند و کاملاً شناخته شده مورد توجه زیست شناسان و محققان رشته پزشکی میباشد. در سال2002، زمانی که مجله Science کشف پدیده تداخل RNA را بزرگترین پیشرفت علمی سال نامید، محققان دریافتند که این کشف ارزشمند علمی طی سالهای آینده چهره تحقیقات حوضه زیستپزشکی (Biomedical research) را متحول خواهد ساخت.
مکانیسم عمل و زیست شناسی فرآیند تداخل RNA:
تداخل RNA به عنوان جدید ترین و کارآمد ترین ابزار خاموش کردن اختصاصی بیان ژنها در سطح پس از نسخهبرداری
(Post-tarnscriptional level) شناخته شده است و در حال حاضر بهترین روش جهت مطالعهی معکوس ژنتیک
(Reverse genetics) در مقایسه با روشهای قدیمیتر خاموش کردن ژنها میباشد. نتایج تحقیقات زیادی در این زمینه نشان میدهد که این روش بسیار اختصاصی عمل میکند و راهاندازی چنین سیستمی در قیاس با سایر سیستمهای غیر فعالسازی ژنها سادهتر و ارزانتر است.
برای مکانیسم عمل فرآیند تداخل RNA یک مدل دو مرحلهای ارائه شده است که شامل دو مرحلهی شروع (Initiation step) و مرحلهی عملکننده (Effector step) است. در مرحلهی شروع مولکولهای RNA دورشتهای به طول حدوداً 500-200 جفت باز توسط فعالیت ریبواندونوکلئازی آنزیمهایی از خانواده RNase III به نام Dicer (DCR) و Dicer-like، با مصرف ATP به قطعات کوچک 23-21 نوکلئوتیدی به نام RNAهای کوچک یا کوتاه مداخلهگر
(small or short interfering RNA) و یا siRNA که به عنوان RNAهای راهنما (guide RNA) عمل خواهند کرد، شکسته میشوند. اعضای خانواده آنزیمهای RNase III جزء معدود نوکلئازهایی هستند که فعالیت اختصاصی برای مولکولهای dsRNA دارند و آنها را در انتهای 3’ به گونهای برش میدهند که 3-2 نوکلئوتید بطور آویزان (overhang) باقی میماند و انتهاهای 5’ و 3’ هیدروکسیله ایجاد میکنند. این نوکلئازها از نظر تکاملی در کرمها، مگس سرکه، قارچها، گیاهان و پستانداران کاملاً حفظ شدهاند. سپس در مرحلهی عملکننده هر کدام از قطعات siRNA در یک مجموعه پروتئینی به نام کمپلکس خاموشکننده القاء شده توسط RNA (RNA-induced silencing complex) یا RISC وارد میشوند. سپس RISC با فعالیت هلیکازی وابسته به ATP، دو رشته siRNA را از هم باز میکند. در این مرحله RISC فعال شده و به همراه مولکول siRNA تک رشتهای که دراین هنگام به آن antisenseRNA نیز میگویند و مکمل ناحیهای بر روی مولکول RNA هدف (target RNA) و یا senseRNA میباشد، به سمت mRNA هدف راهنمایی میشود. سپس زیر واحد اندوریبونوکلئازی موجود در ساختار RISC، mRNA هدف را از وسط میبرد و در نهایت mRNA بریده شده (احتمالاً بوسط اگزوریبونوکلئازها) بطور کامل تخریب میشود و سبب خاموش شدن بیان ژن هدف میشود.
منابع مختلف dsRNA جهت شروع فرآیند RNAi:
همانطور که میدانیم حضور مولکولهای RNA دو رشتهای بلند جهت شروع فرآیند تداخل RNA مهم و حیاتی میباشد. این مولکولهای dsRNA از منابع ذیل و به طرق مختلف میتوانند وارد سلول شده و فرآیند تداخل RNA را راهاندازی کنند:
1- ممکن است از روی لوکوسهای ژنی بازآرایی شده (Rearranged loci)رونویسی شوند.
2- از طریق نسخهبرداری ژنهای دارای پروموتورهای همگرا (Converging promoters).
3- توسط تزریق ترانسژنهای با توالی تکراری معکوس (Inverted repeat transgenes)، که ابتدا در هستهی سلول به صورت مولکولهای RNA دو رشتهای سنجاقسری (hairpin dsRNA) نسخهبرداری شده و پس از ورود به سیتوزول به dsRNA تبدیل میشوند.
4- از طریق نسخهبرداری از روی عناصر قابل انتقال همچون ترانسپوزونها که در ژنوم اکثر سلولهای یوکاریوتی وجود دارند و میتوانند نسخههایی از ژنها را برای هر دو رشته تولید کنند. این رشتهها به صورت سنس و آنتیسنس رونویسی شده و سپس به یکدیگر اتصال یافته تا مولکول dsRNA تشکیل دهند.
5- توسط رونویسی از روی mRNA هدف بوسیلهی آنزیم RdRP (RNA-dependent RNA Polymerase) میزبان یا ویروس.
6- بعضی از ویروسهای حاوی RNA و حتی حاوی DNA طی مراحل تکثیرشان به طور طبیعی میتوانند مولکول RNA دو رشتهای تولید کنند.
7- میتوان مولکولهای dsRNA را به طور سنتتیک در خارج از سلول تولید و سپس به طرق مختلف به سلول تزریق کرد (Injection of synthetic dsRNA).
8- گاهی نیز طی فرآیندی به نام تداخل RNA گذرا (Transitive RNAi) توسط آنزیم RdRP که در برخی از سلولهای گیاهی و قارچها وجود دارد، از روی mRNAهای هدف نسخهبرداری شده و مولکولهای siRNA ثانویه
siRNA) (secondary تولید میشوند.
9- تولید مولکولهای siRNA به وسیلهی ورود ناقلهای DNA همچون پلاسمیدهای باکتریایی و وکتورهای ویروسی (retrovirus and adenovirus-based systems) به درون سلول.
سیستمهای انتقالی و تحویلدهنده siRNA به درون سلول :(Transfection and delivery systems)
از روشهای مختلفی جهت انتقال مولکولهای siRNA به درون سلولها استفاده میشود. مهمترین و رایجترین روشهای انتقال شامل موارد ذیل میباشد:
1- با استفاده از یکسری محلولهای شیمیایی به نام معرفهای آلودهکننده (Transfection reagents)، همچون Lipofectamine 2000، Oligofectamine، TransIT-TKO (Mirus)، Ambion’s Siport Amine and Siport.
2- با استفاده از روش الکتروپوراسیون (Electroporation)
3- تزریق به سلول با روش Microinjection
4- از طریق تجویز داخل وریدی کپسولهای لیپیدی حاوی siRNA (Lipid-encapsulated siRNA)
5- تحویل و هدایت siRNA به سلول توسط آنتیبادیهای اختصاصی نوع سلول (Antibody-directed)
6- با استفاده از سیستمهای تحویلدهنده بر پایه فنآوری نانو و ذرات نانو(Nanoparticle-based siRNA delivery)
کاربردهای تحقیقاتی و درمانی فرآیند تداخل RNA:
با ظهور دانش زیستشناسی مولکولی، عملکرد بسیاری از ژنها با مطالعه فنوتیپ ژنهای جهشیافته (Forward genetics) تا حدی مشخص میشد و سر نخی از عملکرد ژن مورد نظر به دست میآمد. اما با اختراع پروژههای تعیین توالی در مقیاس گسترده، هزاران ژن در موجودات گوناگون بدون اینکه عملکردشان مشخص باشد شناسایی شدند. امروزه با استفاده از دانش ژنتیک معکوس، که در حال حاضر کارآمدترین روش ارزیابی نقش و عملکرد ژنها میباشد، عملکرد بسیاری از ژنهای تعیین توالی شده، مشخص شده است. چندین روش مولکولی که در ژنتیک معکوس جهت هدف قرار دادن ژنها مورد استفاده قرار گرفتهاند عبارتند از: روش نوترکیبی همسان (Homologous recombination) که روشی وقتگیر و گران است، استفاده از الیگونوکلئوتیدهای آنتیسنس (antisense oligonucleotides) و فنآوری رایبوزیم (Ribozyme technology). این روشها علیرغم اینکه در دانش ژنتیک معکوس مفید بودهاند اما دارای محدودیتهایی نیز میباشند. اما با ظهور فنآوری تداخل RNA و بکارگیری RNAهای کوچک مداخلهگر یا همان siRNA جهت خاموش کردن بیان هر ژنی
technonlgy) knockdown (Gene، انقلاب بزرگی در دانش ژنتیک معکوس حاصل شده است.
بر طبق آخرین تحقیقاتی که در زمینه منشاء بسیاری از بیماریها از جمله اختلالات التهابی و سرطانهای خاص انجام گرفتهاست، اساساً اغلب این بیماریها و ناهنجاریها، منشاء ژنی (gene-based) دارند. برای مثال در درمان بسیاری از سرطانها از جمله سرطان روده بزرگ (colon cancer)، سرطان سینه، انواع کارسینوماها، انواع لوسمیها و سرطان پانکراس، با سرکوب بیان ژن و یا ژنهای دخیل در ایجاد بدخیمی توسط فرآیند تداخل RNA، موفقیتهایی حاصل شده است. همچنین در درمان بسیاری از عفونتهای ویروسی و انگلی نیز با بکارگیری سرکوب بیان ژن در سطح پس از نسخهبراری با استفاده از مولکولهای siRNA، از تکثیر و پیشرفت عفونت جلوگیری به عمل آمده است. برای مثال با سرکوب ژن Vif که یک ژن تنظیمی در ویروس HIV-1 میباشد، از همانندسازی و تکثیر ویروس ممانعت به عمل آمده است. علاوه بر این در درمان برخی از عفونتهای باکتریایی همچون پنومونی و شوک عفونی (septic shock) نیز از این تکنیک با موفقیت استفاده شده است (جدول 1).
جدول 1- برخی از ژنهای ویروسی که توسط siRNA مورد هدف قرار گرفتهاند.
کاربرد فرآیند RNAi در درمان عفونتهای باکتریایی:
پروکاریوتها خصوصاً باکتریها به دلیل اینکه اکثراً خارج سلولیاند و در خارج سلول تکثیر پیدا میکنند و فاقد مکانیسم RNAi میباشند، اصولاً تحت تاثیر خاموشی ژنها توسط فرآیند تداخل RNA قرار نمیگیرند. اما این امکان وجود دارد که بتوان با سرکوب بیان آن دسته از ژنهایی که در تحریک بیش از حد سیستم ایمنی منجر به پاسخهای ناخواسته میشوند، میزان مرگ و میر عفونتهای باکتریایی را کاهش داد. برای مثال میتوان با سرکوب بیان ژنهای مسئول در تولید سایتوکینهای پیشالتهابی و التهابیcytokines) (pro-inflammatory and inflammatory، همچون IL-1 و TNF-α که در ایجاد شوک عفونی نقش بسزایی دارند، پاسخ ایمنی میزبان را کنترل کرد بدون اینکه در توسعهی پاسخ ایمنی حفاظتی خللی ایجاد شود. در مطالعهای که در سال 2006 توسط Yanagihara و همکاراناش در ژاپن انجام شد این محققان توانستند با استفاده از مولکولهای اختصاصی siRNA تا حدود زیادی موجب سرکوب بیان ژن مسئول در تولید آنزیم کوآگولاز استافیلوکوکوس اورئوس مقاوم به متیسیلین (MRSA) شده و قدرت آسیبزایی این باکتری را در یک مدل موشی عفونت ریوی به شدت کاهش دهند.
در خاتمه بایستی اذعان کرد که علاوه بر کاربردهای دیگر فرآیند تداخل RNA در جنبههای مختلف تحقیقات پایه از جمله ژنومیک عملکردی (Functional genomics) و تایید اهداف دارویی جدید (Drug target validation)، فنآوری سرکوب بیان ژن توسط siRNA میتواند به عنوان یک گزینه درمانی کارآمد جهت پیشگیری و مهار پروسههای بیماریزایی عفونتهای میکروبی خصوصاً عوامل بیماریزای مقاوم به چند دارو مورد توجه روز افزون قرار گیرد.
منبع : www.kafil.blogfa.com/
ساختمان DNA
ساختمان رشتهای DNA
سرعت پیشرفت تعیین ساختمان DNA بسیار کند بوده است. در سال 1930 کاسل و لویننوکلئوتید میباشد که از سه قسمت تشکل شده است. یک قند پنتوز (2- دزوکسی D- ریبوز) ، یک گروه 5-فسفات و از یکی چهار باز آلی نیتروژندار حلقوی آدنین (A) ، گوانین (G) ، سیتوزین (C) و تیمین (T) تشکیل شده است.
دریافتند که نوکلئین در واقع اسید دزوکسی ریبونوکلئیک است. برسیهای شیمیایی آن مشخص کرد که زیر واحد تکرار شونده اصلی DNA ،از این چهار باز دو باز آدنین و گوانین از بازهای پورینی و دو باز سیتوزین و تیمین از بازهای پیریمیدینی میباشند. به مجموعه قند و باز آلی نوکلئوزید گفته میشود. گروه فسفات میتواند به کربن3 و یا5 متصل شود. به مجموع نوکلئوزید و گروه فسفات متصل به آن نوکلئوتید میگویند. با توجه به اینکه یون فسفات میتواند هم به کربن 3 و هم به کربن5 متصل شود.
پس دو نوکلئوتید از طریق یک پیوند فسفودی استر بهم متصل میشوند. به این صورت که گروه هیدروکسیل یک نوکلئوتید با گروه فسفات نوکلئوتید دیگر واکنش داده و پیوند فسفودی استر را بوجود میآورد. از آنجایی که پیوند فسفودی استر ، کربنهای3 و5 دو قند مجاور را بهم متصل میکند، این پیوند را پیوند5-3 فسفودی استر نیز مینامند. یک زنجیره در اثر اتصال پشت سر هم تعدادی2-دزوکسی ریبونوکلئوتید بوسیله پیوندهای دزوکسی ریبونوکلئوتید تشکیل میشود.
تمامی نوکلئوتیدها در یک زنجیره پلی نوکلئوتیدی دارای جهت یکسان میباشند. به این صورت که نوکلئوتید انتهایی در یک سمت زنجیره دارای یک گروه5 آزاد و نوکلئوتید انتهایی در سمت دیگر زنجیره دارای یک گروه3 آزاد میباشد. بنابراین زنجیره پلی نوکلئوتیدی دارای جهت بوده و این جهت را به صورت5--->3 نشان میدهند. بنابراین اگر در نوکلئوتید ابتدایی کربن5 در بالای حلقه پنتوز و کربن3 در زیر آن باشد، در تمامی نوکلئوتیدهای بعدی زنجیره کربن 5 در بالای حلقه پنتوز جای خواهد داشت.
نتایج حاصل تا سال 1950
- DNA یک پلیمر رشتهای متشکل از واحدهای2- دزوکسی اسید ریبونوکلئیک میباشد که بوسیله پیوندهای فسفودی استر5-3 به هم متصل شدهاند.
- DNA حاوی چهار زیر واحد dc و dG و dT و dA میباشد.
- مقادیر متوالی dT و dA با یکدیگر و dc و dG نیز با یکدیگر مساوی میباشند.
مارپیچ دو رشتهای DNA
در سال 1953 در ساختمان سه بعدی DNA ، بوسیله واتسون و کریک کشف شد. واتسون و کریک با استفاده از مطالعات تفرق اشعه ایکس ، رشتههای DNA که بوسیله فرانکلین و ویلکینز
تهیه شده بود و همچنین ساختن مدلها و استنباطهای مشخصی ، مدل فضایی خود را ارائه دادند و در سال 1962 واتسون و کریک و ویلکینز به خاطر اهمیت کشف ساختمان DNA به صورت مشترک جایزه نوبل دریافت کردند.مدل پیشنهادی آنان چنین بود. DNA یک مارپیچ دو رشتهای است که رشتههای آن به دور یک محور مرکزی ، معمولا به صورت راست گرد پیچ میخورند. طبق مدل واتسون و کریک ، ستونهای قند - فسفات همانند نردههای پلکان به دو قسمت خارجی بازهای آلی پیچیده و به این ترتیب در معرض محیط آبکی داخل سلول هستند و بازهای آلی که خاصیت آبگریزی دارند، در داخل مارپیچ قرار میگیرند. هنگام تشکیل مارپیچ رشتهها به صورت موازی متقابل قرار میگیرند.
یعنی اگر جهت یک رشته3<--5 باشد، رشته دیگر 5<--3 خواهد بود. پیوندهای هیدروژنی بین آدنین از یک رشته با باز تیمین رشته مقابل و باز گوانین یک رشته با سیتوزین رشته مقابل بوجود میآیند. گر چه از نظر اندازه هر باز پورینی میتواند در مقابل یک باز پیریمیدین قرار بگیرد. ولی به دلیل وجود گروههای شیمیایی روی بازهای G و C و T و A پیوندهای هیدروژنی مناسب فقط بین C - G و T - A برقرار میشود و ایجاد پیوند بین T - G و C- A ممکن نیست.
واکنشهای توتومریزاسیون
اتم هیدروژن در بازهای آلی میتواند روی اتمهای نیتروژن و یا اکسیژن حلقه جابجا شود. این تغییر موقعیت هیدروژن روی حلقه باز را توتومریزاسیون میگویند. توتومریزاسیون در بازهای آدنین سیتوزین باعث تبدیل فرم آمینی به فرم ایمنی و در مورد بازهای تیمین و گوانین باعث تبدیل فرم کتونی به فرم انولی میشود.
در شرایط فیزیولوژیکی ثابت تعادل واکنش توتومریزاسیون بیشتر به سمت اشکال آمینی و کتونی میباشد. این حالت پایدار پروتونی ، الگوی تشکل پیوندهای هیدروژنی بین بازها را تعیین مینماید، بطوری که بازهای T و A با تشکیل دو پیوند هیدروژنی و بازهای G و C با سه پیوند هیدروژنی با هم جفت میشوند. C و A و همچنین T و G نمیتوانند با هم جفت شوند.
زیرا در این بازها اتمهای هیدروژن هر دو در یک موقعیت قرار دارند و امکان ایجاد پیوند هیدروژنی وجود ندارد. به دلیل اینکه در رشتههای DNA همواره باز A مقابل T و باز G مقابل C قرار دارد، این دو رشته را مکمل مینامند. بنابراین توالی موجود در یک رشته DNA ، توالی رشته مقابل را تعیین میکند. مکمل بودن دو رشته DNA ، اساس عمل همانند سازی DNA است
تهیه ی بلور DNA
مقاله ای با عنوان زیر در مورد تهیه بلور DNA جلب توجه نمود:
به منظور بررسی شیار اصلی در Z-DNA.بلور های کوچک از یک اولیگونوکلئوتید سنتتیک تهیه و به وسیله انتشار بخار(Vapor Diffusion ) با استفاده از دو روش قطره آویزان(hanging drop method ) و قطره نشسته(sitting drop method ) بلور ها رشد داده شدند و با قرار دادن بلور ها در لوله های سیلیکون و با استفاده از مطالعات نوترونی به بررسی بلور ها پرداختند.*
برای تهیه بلور از یک ماده چند کار بایستی انجام گیرد 1-خالص سازی آن ماده وتهیه بلور کوچک از آن ماده
2-رشد بلور آن ماده
برای خالص سازی یک ماده از روش های متعددی استفاده می شود .
جذب سطحی –سانتریفیوژ -کروماتوگرافی-بلور سازی-تبخیر-تقطیر-خشک کردن-الکتروفورز-تبلور مجدد-رسوب سازی-یخ زدن جز به جز-تقطیر جز به جز-صاف کردن-تصعید- و...در انواع گوناگون روند های جداسازی مخلوطی از ماده اصلی و ناخالصی وجود داردکه در مقادیری از حلال حل شده اند.**

روش بسیار شایع برای بلور سازی استفاده از تبخیر(Vapor Diffusion ) می باشد.این روش خود به دو صورت قطره آویزان(hanging drop method ) و قطره نشسته(sitting drop method ) قابل اجرا می باشد.در هر دو روش نیاز به یک قطره حاوی آن ماده خالص(پروتئین یا DNA) وبافر و رسوب دهنده میباشد این مواد بایستی با
یک ظرف بزرگ حاوی بافر های مشابه و رسوب دهنده مشابه در غلظت های بسیار
بالاتر به حالت تعادل برسند.در ابتدا قطره حاوی آن ماده دارای غلظت مناسبی
برای بلور سازی نمی باشد اما همانطور که آب از قطره بخار
می شود و وارد ظرف می شود غلظت ماده راسب اقزایش می یابد تا به یک سطح
مناسب برای بلور سازی برسد. از آنجائی که سیستم در حال تعادل است این
حالات مناسب حفظ می شود تا زمانی که بلور سازی کامل گردد.تفاوت دو روش
قطره آویزان و قطره نشسته در جهت گیری قرار دادن قطره حاوی آن ماده در
سیستم می باشد.هر دو روش نیازمند یک سیستم بسته ای هستند سیستمی
که بایستی کاملا از هوای بیرون ایزوله گردد این کار به وسیله ظرف غیر قابل
نفوذ و یا گریس باقدرت خلاء بالا بین سطوح قابل اجرا می باشد.***
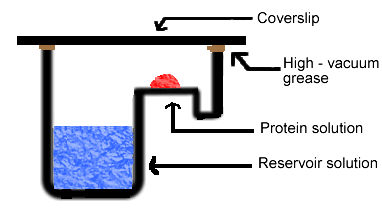
روش قطره نشسته(sitting drop method )
در این روش قطره حاوی آن ماده(پروتئین) بر روی یک ستون و در بالای ظرف محلول قرار دارد(بر خلاف روش قطره آویزان).
http://scripts.iucr.org/cgi-bin/paper?S174430910601236X
http://en.wikipedia.org/wiki/Separation_process
http://en.wikipedia.org/wiki/List_of_purification_methods_in_chemistry
http://www.bio.davidson.edu/COURSES/Molbio/MolStudents/spring2003/Kogoy/protein.html
SORCE:
http://www.groupbio.blogfa.com/post-257.aspx
از اندوسیتوز تا اگزوسیتوز
آندوسیتوز
اکثر مواد از طریق انتشار یا انتقال فعال به درون سلول راه مییابد. اما ذرات بسیار بزرگ بواسطه یک عمل تخصص یافته غشای سلولی موسوم به آندوسیتوز وارد سلول میشود. اشکال اصلی آندوسیتوز عبارتند از پینوسیتوز (قطره خواری) و فاگوسیتوز (ذره خواری).

پینوسیتوز
در زیر گودهها و بر روی سطح درونی غشای سلول ، شبکهای از یک پروتئین فیبریلی به نام کلاترین و نیز پروتئیهای دیگر وجود دارد که احتمالا شامل فیلمانهای انقباضی اکتین و میوزین میباشد. هنگامی که مولکولهای پروتئینی به گیرندهها متصل میشود خصوصیات سطح غشا چنان تغییر میکند که تمام گوده به درون سلول فرو میرود و پروتئینهای پیرامون آن (اکتین و میوزین) تدریجا به هم نزدیک میشوند و لبههای گوده به هم میرسند. بلافاصله پس از آن ، قسمت فرو رفته غشا از سطح سلول جدا میشود و یک وزیکول پینوسیتوزی در سیتوپلاسم سلول بوجود میآید. انرژی این عمل از مولکول ATP تامین میشود و ضمنا برای این عمل وجود یون کلسیم در مایع خارج سلولی ضرورت دارد.
فاگوسیتوز
در مورد باکتریها ، معمولا باکتری قبلا به این آنتیبادی خاصی متصل شده است و اتصال آنتیبادی به گیرندههای فاگوسیت موجب کشیده شدن هر دوی آنها به درون فاگوسیت میشود. به این عمل واسطهای آنتیبادیها اپسونیزاسیون گویند.
اگزوسیتوز
خروج مواد بزرگ یا مولکولهای درشت از سلول و ورود آن به مایع خارج سلولی توسط پدیده اگزوسیتوز انجام میگیرد. معمولا باقیمانده مواد هضم شده توسط لیزوزومها که دیگر قابل تجزیه نیستند و به جسم باقیمانده معروف هستند توسط عمل اگزوسیتوز از سلول خارج میشود. عمل اگزوسیتوز درست عکس عمل اندوسیتوز میباشد.